|
||||
Getting the Most Out of the Protein Data Bank |
David S. Goodsell The Protein Data Bank archive (PDB) is the primary repository for atomic structures of biologically important molecules. These structures are available for use free of charge, and thus provide an invaluable resource to illustrators and educators in the biological sciences. The structures, however, pose many challenges for the non-expert user. Several of the major challenges are presented here along with ideas for how approach them.
|
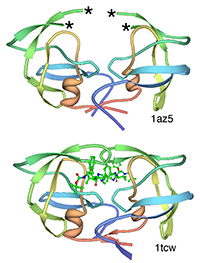
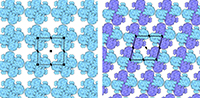
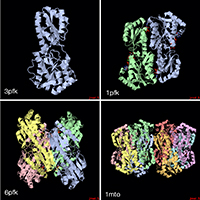
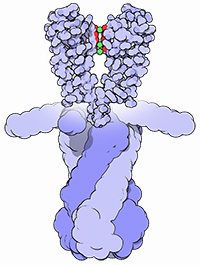
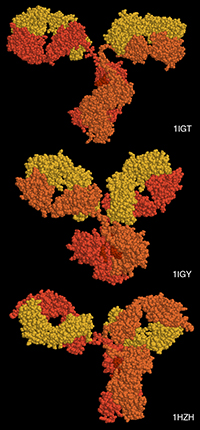
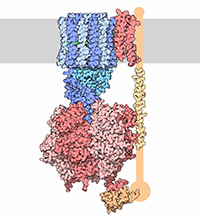
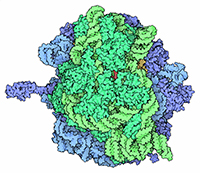
With the steady growth of knowledge in molecular biology and its application to health, scientific illustrators are increasingly being called upon to create images of molecular subjects. In many cases, the structures of biologically important molecules are now known, and are made available for use through the Protein Data Bank. The resource, however, is designed with structural biologists in mind, and it is often difficult for non-experts to find and use the atomic information that they need. The Protein Data Bank continues to improve the tools for searching, exploring, and downloading the 60,000 structures housed in the database, but there are still many pitfalls waiting to trap the unwary user. This paper presents several of the most common challenges, and when possible, suggest some solutions. Introduction to the PDB The Protein Data Bank was created in 1971 at the Brookhaven National Laboratory as a way to archive results from the new field of structural molecular biology (Bernstein et al. 1977). At the time, there where seven atomic structures of proteins and the coordinates were distributed on magnetic tape to interested users. In 1998, the Research Collaboratory for Structural Biology (RCSB) took over management of the PDB (Berman et al. 2000). They responded with updates to the underlying computational structure of the database, an improved WWW site, and comprehensive system for validating and annotating structures submitted to the database. PDB data are created and used throughout the world, so the Worldwide Protein Data Bank (http://www.wwpdb.org) was established in 2003 to ensure that the archive is freely and publicly available to the global community (Berman et al. 2003). The wwPDB members (the RCSB PDB and BioMagResBank in the United States, PDBe in Europe, and PDBj in Japan) host deposition, processing, and distribution centers for PDB data and collaboration on a variety of projects and outreach efforts. Most recently, the wwPDB performed a remediation of the entire database to ensure consistency of files throughout. Today, the PDB is the single worldwide repository of information about the 3D structure of large biological molecules, such as proteins and nucleic acids. Most of the structures are obtained from x-ray crystallography, but the archive also includes structures determined by NMR spectroscopy, electron microscopy, and other techniques. Most users will access the RCSB PDB through its convenient WWW site (http://www.pdb.org). The homepage includes a search window, as well as many informational and news resources. Typing a molecule name in the search window will yield a list of one or more entries, and clicking on an entry leads to the Structure Summary page. Many types of information are available on this page, including basic information on the structure (title, author, description, etc), tools for display of the structure, including a static image and links to several interactive viewers, and menus for downloading the files, including the coordinate files used by most molecular illustration programs. The Structure Summary page also includes a series of tabs that lead to annotation information for the entry. These include pages to explore the amino acid sequence, to find homologous structures in the PDB, a literature page, and pages on the biology and crystallographic details. The RCSB PDB WWW site includes tutorials and help services for all aspects of the archive. Several introductory resources are available for new users, including “Looking at Structures” (a more detailed presentation of the material in this paper), “Molecule of the Month” (which regularly highlights the structural and functional significance of different biological molecules), and other educational resources. Pitfalls: Molecules in CrystalsMany difficulties encountered with PDB files are caused by the underlying science of crystallography. To solve a crystal structure, a concentrated solution of the molecule is used to grow a crystal, which is then subjected to an intense beam of x-rays. The beam is diffracted into a characteristic pattern of spots, which are then used to create an image of all of the electrons in the crystal. This image is then interpreted as the collection of atoms that make up the molecule. The PDB typically stores the data for the diffraction pattern, and the list of atomic coordinates. These coordinates are most often used to create illustrations of the molecules. Unfortunately, many challenges are caused by the fact that these molecules are observed in the context of a crystal. 1) Where are all the hydrogen atoms? The crystallographic technique observes the electrons in molecules. Since hydrogen atoms have only a single electron, they do not provide enough scattering power to be seen, except in rare highly ordered crystals. So, atomic coordinates of hydrogen atoms are not usually included in PDB file, since they are not observed experimentally. Fortunately, coordinates for hydrogen atoms are easily calculated based on the known geometry of biological molecules. Free programs such as Reduce (Word et al. 1999) may be used to create the coordinates. 2) My structure is missing some important loops. X-ray crystallography relies on the fact that crystals contain millions of copies of the molecule in exactly the same orientation. However, if there is a flexible loop, it will have different conformations in all the different molecules in the crystal, and the experimental image of the loop will be smeared out. Often, crystallographers do not include coordinates for these loops, since there is not good experimental evidence for their location. In other cases, they may include several versions of the loop. An example from HIV protease is illustrated in Figure 1. This is a tricky problem to solve. Researchers in biomolecular structure typically use molecular modeling programs (which allow the user to build new coordinates based on the known molecular geometry) to create a hypothetical model of missing regions. One possible solution is to continue looking in the PDB for other structures—in many cases, addition of inhibitors or substrates will lock down the conformation of a flexible loop, so a structure with an inhibitor may be the better choice for creating an illustration. 3) The file only contains one subunit, and I know the protein contains four. One of great joys of being a crystallographer is the wonderful symmetry of the crystalline state. Unfortunately, this symmetry can cause complications when you go to use the molecules for a non-crystallographic application, such as an illustration. Problems occur when a symmetrical molecule is crystallized, and the symmetry of the crystal matches the symmetry of the molecule (Figure 2). In these cases, only the unique portion of the molecule, termed the “asymmetric unit,” is included in the PDB file, since the user can generate the functional form of the molecule, termed the “biological assembly,” using the symmetry transformations of the crystal. An example is included in Figure 3. Fortunately, atomic coordinates for the functional biological assemblies for all entries are available from the PDB archive. There is one important caveat to this, however: the assemblies are necessarily limited to the assemblies that are actually present in the crystal. So, the biological assembly provided by the PDB will typically have the desired assembly for oligomeric proteins like hemoglobin or catalase, but biological assemblies for large structures like actin filaments or microtubules are not provided by the PDB since these assemblies are not formed in the crystal lattice. 4) The structure only includes alpha carbon atoms. In some cases, the quality of crystals may be poor, so the researcher interprets the diffraction pattern to give only the overall fold of the protein chain instead of positions for every atom. For these structures, the PDB file will only include one atom per amino acid, typically the alpha carbon position. These types of files are also occasionally seen for structures solved with electron microscopy, which typically does not have the power to resolve atoms. With these structures, of course, you are stuck. You can create ribbon diagrams with this data, and by using large spheres (4.5 – 5 Å) you can approximate the molecular surface (Figure 4). But in order to create atomic-level images, you need to model all the atoms that are missing, or go back to the PDB and see if there is another structure with more complete coordinates. 5) Crystal contacts are perturbing the structure. The molecules in crystal structures are packed tightly together, and often they bend and distort their neighbors. This can be seen in a series of antibody structures (Figure 5). These structures are composed of three large domains connected by flexible linkers. Looking at the different structures, we see a variety of crazy conformations as the three domains find their own places in the crystal lattice. This underscores the fact that crystallographic structures are just a single snapshot of the molecule—for flexible molecules like antibodies, there may be many other (perhaps more aesthetically pleasing) conformations. So in these cases, it pays to be critical when you’re searching for an appropriate PDB file for a project. Pitfalls: the Games Crystallographers PlayThe crystallization of biological molecules is a fine art, and in many cases, luck and serendipity play a big role. Crystallographers do many things to their molecules to assist the process of obtaining crystals, and this is often reflected in the atomic coordinates that they deposit in the PDB. 1) This protein is from E. coli, and I need the human protein. Crystallographers typically solve the structure of whatever form crystallizes. In many cases, they will try to use the protein from a number of different organisms, hoping that one of them will crystallize. In recent years, this is not as necessary, since crystal screening and robotics have automated the process and increased the odds that a crystal may be obtained for the desired molecule from the desired organism. But when you go to the PDB looking for a structure, you realize that the structure may not be available from the organism you desire, but may be isolated from a related organism. The RCSB PDB WWW site has tools for looking for related proteins in the “Sequence Similarity” tab for each PDB entry. 2) The PDB file only contains part of the protein. For very flexible proteins, crystallographers often cut the protein into manageable pieces, and solve the structure of the individual parts. For many years, the structures of the isolated Fc and Fab domains were the only structures available for antibodies—only recently have crystals been obtained of intact antibodies. ATP synthase (Figure 6) is another example—the PDB archive does not currently hold a structure of the entire complex, but structures of various fragments are available. Unfortunately, this is a difficult problem to solve as an illustrator. The PDB files will usually report that the structure is a fragment of the whole protein (sometimes you have to read carefully to determine this), but it will rarely tell you if structures of the other pieces are available. In most cases, a careful reading of the scientific literature is needed to assemble the necessary pieces for a complete image. Then, the user is faced with the same challenge as with missing loops: the user must assemble the pieces and generate hypothetical coordinates for the missing portions. 3) What are all these histidines and selenium atoms? Crystallographers are always looking for ways to make their jobs easier, so they can solve more and more structures. Two common techniques are widely used to simplify the crystallographic structure determination, and they often leave footprints in the PDB files. The first is a histidine tag—this is a string of histidine amino acids that are engineered into the beginning or end of the protein. These are used in a rapid one-step method for purifying the protein. Fortunately, this little tail of histidines is usually very flexible and is disordered in the crystals, so coordinates are rarely included. Figure 7 shows a case where the histidine tag was seen in the crystal. A second common modification is the incorporation of selenium atoms in methionine amino acids, in place of the normal sulfur atoms. These selenium atoms diffract x-rays in an unusual way that simplifies the calculation of the initial image of the electron density, and thus they are used in many current structures (Figure 7). Fortunately, selenium is very similar in chemical characteristics to sulfur, and often may be treated as a sulfur atom for images. 4) Where is the glycosylation? Many proteins on the surfaces of cells and viruses are decorated with carbohydrate chains. For instance, the GP120 protein on the surface of HIV is about half carbohydrate and half protein. Unfortunately, these carbohydrate chains are very flexible, and thus glycoproteins are difficult to crystallize. So, crystallographers often use specific enzymes to clip off the carbohydrate chains, and they crystallize the deglycosylated molecules. In many cases this is not a problem since the carbohydrate chains do not play a specific role in the function. In cases where they are important, however, coordinates for the carbohydrate chains may be built using a molecular modeling program. 5) The substrate looks odd. Crystallographic experiments typically take days or weeks to perform, so the molecules used in the crystal must be stable for this amount of time. This is tricky when you’re trying to solve the structure of an enzyme with its natural substrates: if you try to grow a crystal, the enzyme will simply do its job and convert all the substrate into products, and you won’t see anything. To solve this problem, crystallographers use molecules that are similar to the substrates, but have small changes that make them resistant to the action of the enzyme. For instance, Figure 7 shows a myosin structure with an analogue of ATP that cannot be cleaved by the motor protein. Nearly every file in the PDB that includes small molecules will have this type of modification, using analogues or mimics or inhibitors instead of the natural substrates. So, when you get a file from the PDB, you must be critical and carefully use this as a model for the real substrate. Pitfalls: PDB-ismsA final problem is caused by the underlying structure of the PDB file format. The atom number record is formatted such that only 99,999 atoms may be included in the file. Amazingly, many current structures exceed this limit. Most notably, the structures of whole ribosomes (Figure 8) have over 140,000 atoms. The wwPDB solves this by splitting the coordinates into several files. Thus, to create a picture you need to recombine the files. Many molecular graphics programs allow the user to read several files simultaneously, but I usually find it easier to append the files using a text editor. Problems occasionally occur with the names of the individual subunits—if similar names are used in the two separated files (such as chain “A”, chain “B”, etc), there will be ambiguity when the files are combined, and it is left for the display software to provide a way to specify each. How Do I Choose a File? This is arguably the most challenging aspect of using the PDB archive: how do you find a suitable entry in a database of 60,000 structures? The Molecule of the Month series is intended to assist with this challenge. In each installment, I have identified a few PDB entries that exemplify particular functional features for popular molecules, so it often provides an easy place to start. As you enter the database to search for an appropriate entry, there are many things to take into consideration, and fortunately, the PDB WWW site has many tools for sorting through these possibilities. A simple keyword search will typically yield 10-100 possibilities for a given protein. I typically use two basic criteria when choosing a particular file: 1) The resolution. Resolution is a measure of the quality of the crystal, and thus of the quality of the structure. Lower numbers correspond to structures with more data, and thus better levels of accuracy. Structures solved at 1 Å resolution are among the best structures, 2-3 Å resolution are typical, and structures with greater than 4 Å resolution must be treated with some skepticism. 2) The biology. A careful reading of descriptions will lead you to a structure that is closest to your topic of interest. Look for things like the organism, any inhibitors or substrates included in the structure, and whether the structure is a fragment or the whole protein. The PDB also includes many functional complexes (such as DNA-protein complexes or signaling complexes) so a careful search can often yield something really interesting. The wwPDB is an amazing resource that is waiting to be tapped for all manner of educational and artistic applications. It includes atomic structures for many of the most important molecules in cells, including DNA and DNA polymerase, ribosomes performing many of the steps of protein synthesis, viruses that attack plants and animals, all of the enzymes of glycolysis, drug targets for cancer, heart disease and mental illness, and many, many more. Now, when we need to create an illustration of a biological molecule, we can go to the PDB archive and use the actual atomic structure. Acknowledgements I thank Christine Zardecki and Shuchismita Dutta for helpful comments. This work was supported by the RCSB PDB (NSF DBI 03-12718). This is manuscript number 20474 from the Scripps Research Institute.
References H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, P. E. Bourne (2000) The Protein Data Bank, Nucleic Acids Res 28, 235-242. H. Berman, K. Henrick, H. Nakamura (2003) Announcing the worldwide Protein Data Bank, Nat Struct Biol 10, 980. F. C. Bernstein, T. F. Koetzle, G. J. Williams, E. F. Meyer, Jr., M. D. Brice, J. R. Rodgers, O. Kennard, T. Shimanouchi, M. Tasumi (1977) The Protein Data Bank: a computer-based archival file for macromolecular structures, J Mol Biol 112, 535-542 J. L. Moreland, A. Gramada, O. V. Buzko, Q. Zhang, P. E. Bourne (2005) The Molecular Biology Toolkit (MBT): a modular platform for developing molecular visualization applications. BMC Bioinformatics 6, 21. J. M. Word, S. C. Lovell, J. S. Richardson, D. C. Richardson (1999) Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation, J Mol Biol 285, 1735-1747.
David S. Goodsell is an Associate Professor of Molecular Biology at the Scripps Research Institute. He received his Ph.D. from UCLA, where he used x-ray crystallography and computer graphics to study the structure of DNA. He now divides his time between biomolecular research and science education. In his research, he develops new computational tools to study the basic principles of biomolecular structure and function. He is currently employing these tools to search for new drugs to fight drug resistance in HIV therapy. He is author of the Molecule of the Month, a feature at the RCSB Protein Data Bank that presents a new molecule each month, describing its function and role in health and welfare. His illustrated books The Machinery of Life and Our Molecular Nature explore biological molecules and their diverse roles within living cells, and his book Bionanotechnology: Lessons from Nature presents the growing connections between biology and nanotechnology. goodsell@scripps.edu More information may be found at his WWW site at: http://mgl.scripps.edu/people/goodsell.
|
|
Copyright
2010, The Journal of Biocommunication, All Rights Reserved |